This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Background High-intensity physical activity has traditionally been discouraged in patients with hypertrophic cardiomyopathy due to concerns about triggering suddencardiacdeath. However, current guidelines adopt a more liberal stance, and evidence on risk factors for exercise-related suddencardiacdeath remains limited.
This cohort study evaluates the association of late gadolinium enhancement with suddencardiacdeath or relevant associated events among patients younger than 21 years with hypertrophic cardiomyopathy.
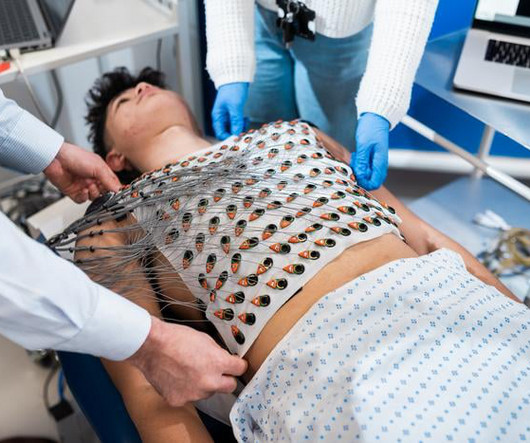
Image courtesy of UCL Institute of Cardiovascular Science / James Tye milla1cf Tue, 12/19/2023 - 18:19 December 19, 2023 — A vest that can map the electrical activity of the heart in fine detail could potentially be used to better identify people at high risk of suddencardiacdeath , suggests a new study led by UCL researchers.
Hypertrophic cardiomyopathy (HCM) is the most common genetic cardiovascular condition. It is the leading cause of suddencardiacdeath in young people and children, with an annual mortality rate of 1%. However, 10% to 20% of these patients have a significantly higher risk of suddencardiacdeath.
This randomized clinical trial assesses whether cardioverter-defibrillator implantation is more effective than amiodarone therapy for the primary prevention of all-cause mortality and secondary prevention of suddencardiacdeath, hospitalization for heart failure, and use of a pacemaker among patients with chronic Chagas cardiomyopathy.
Dilated cardiomyopathy (DCM) is a common heart muscle disorder of nonischemic etiology associated with heart failure development and the risk of malignant ventricular arrhythmias and suddencardiacdeath. Circulation: Genomic and Precision Medicine, Ahead of Print.
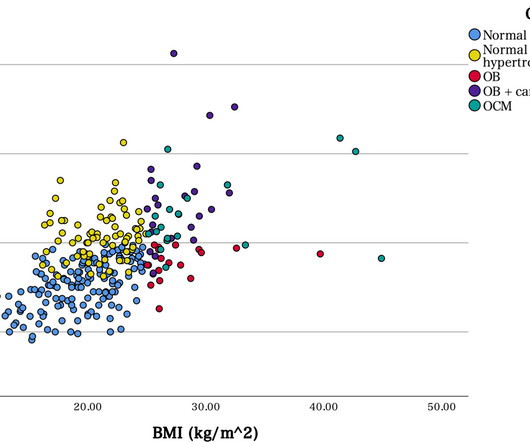
Background We aimed to clarify the existence and pathological features of obesity cardiomyopathy (OCM) in Japan using our series of autopsy cases. Approximately half the OCM cases were diagnosed with suddencardiacdeath (SCD), with significant differences.
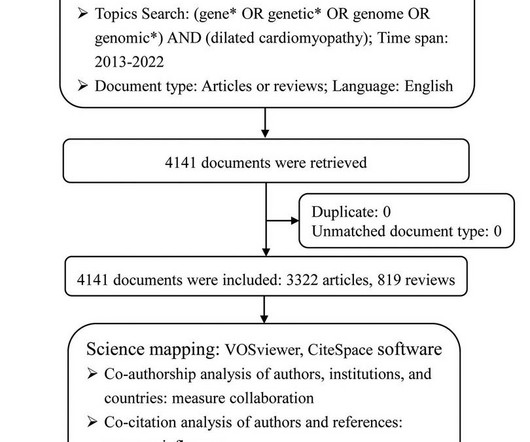
BackgroundDilated cardiomyopathy (DCM) is a heterogeneous myocardial disorder with diverse genetic or acquired origins. Notable advances have been achieved in discovering and understanding the genetics of DCM.
Peripartum Cardiomyopathy (PPCM) is a polymorphic myocardial disease occurring late during pregnancy or early after delivery. The issue is relevant since some arrhythmias are associated to suddencardiacdeath occurring in young patients, and the overall risk does not cease during the early postpartum period.
Suddencardiacdeath represents a significant mortality factor in hypertrophic cardiomyopathy (HCM). Nevertheless, the validation of the existing risk prediction model in the pediatric population remains insufficiently explored.
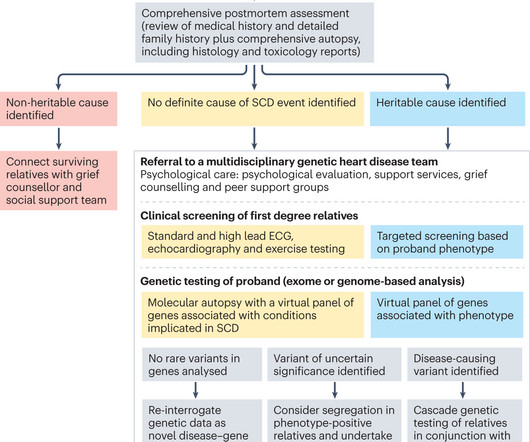
Nature Reviews Cardiology, Published online: 23 January 2024; doi:10.1038/s41569-024-00989-0 A molecular autopsy is undertaken in cases of suddencardiacdeath with no definitive cause found after conventional autopsy, with the aim of identifying a pathological genetic variant that could account for the death.
Objectives To describe a cohort of patients with arrhythmogenic left ventricular cardiomyopathy (ALVC), focusing on the spectrum of the clinical presentations. Twenty-one (41%) had normal echocardiogram, 13 (25%) a hypokinetic non-dilated cardiomyopathy (HNDC) and 17 (33%) a dilated cardiomyopathy (DCM).
Hypertrophic cardiomyopathy (HCM) stands as one of the most common inheritable myocardial disease and is often marked by its association with suddencardiacdeath (SCD) in the young. 1 2 Traditionally, risk assessment in HCM has focused mainly on estimating the likelihood of SCD.
Whether you’re gearing up for your Family Medicine or Cardiology Board Exam, you’ll need to master the topic of Hypertrophic Cardiomyopathy (HCM). It is crucial for every medical physician to recognize patients who are at risk of suddencardiacdeath due to their underlying medical condition.
Maron, MD , a cardiologist and director of the Hypertrophic Cardiomyopathy Center at Lahey Hospital and Medical Center in Burlington, Massachusetts, and the study’s lead author. It causes the heart muscle to become stiff and thick, making it harder for the heart to pump blood properly and increasing the risk of suddencardiacdeath.
Although the 2024 ACC/American Heart Association (AHA) and 2023 European Society of Cardiology (ESC) guidelines stratify well overall the risk of suddencardiacdeath (SCD) in patients with hypertrophic cardiomyopathy (HCM).
Implantable cardioverter defibrillator (ICD) prevents suddencardiacdeath (SCD) in patients with ischemic cardiomyopathy (ICM). Catheter ablation has been shown to effectively reduce ventricular tachycardia (VT) recurrence, yet its efficacy in patients without an ICD implantation remains uncertain.
Suddencardiacdeath (SCD) remains a major health problem in all continents. It usually affects older patients suffering from coronary artery disease and cardiomyopathies. This group of patients defines the sudden arrhythmic death syndrome (SADS) where the cause of death is unknown.
A common characteristic of almost all inherited cardiac disorders is the inherent risk of suddencardiacdeath. Hypertrophic cardiomyopathy (HCM) is one of these conditions and, indeed, risk stratification attempts have been numerous.
The study, titled " Targeted Therapy with Glycogen Synthase Kinase-3 Inhibition for Arrhythmogenic Cardiomyopathy " (TaRGET), will be led by the research team at PHRI and will involve collaboration with multiple organizations in addition to AMO Pharma including the Hearts in Rhythm Organization (HiRO).
What is the prognostic value of extent of late gadolinium enhancement (LGE) in predicting suddencardiacdeath (SCD) in patients with hypertrophic cardiomyopathy (HCM)?
Suddencardiacdeath (SCD) risk stratification is based on clinically recognized risk factors (RF), such as reduced left ventricular (LV) ejection fraction (EF), heart failure (HF), prior myocardial infarction (MI), and syncope. These RFs fail to capture the majority of SCDs.
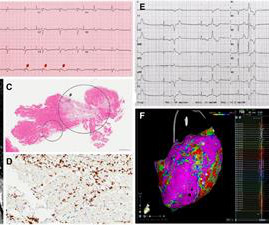
BACKGROUND:Few clinical data are available onNEXNmutation carriers, and the gene’s involvement in cardiomyopathies or suddendeath has not been fully established. years), 21 presented with dilated cardiomyopathy (prevalence, 0.33%), and 3 presented with hypertrophic cardiomyopathy (prevalence, 0.14%).
The American College of Cardiology (ACC) and the American Heart Association (AHA) today released a new clinical guideline for effectively managing individuals diagnosed with hypertrophic cardiomyopathy (HCM). Ommen, MD, FACC , medical director of the Mayo Hypertrophic Cardiomyopathy Clinic and chair of the guideline writing committee.
To improve arrhythmogenic cardiomyopathy (ACM) patient care four pillars of ACM research are necessary. The first pillar to improve ACM is cohort studies in which the genotype-specific natural history of disease is described, and subsequently genotype-specific risk calculators can be developed, such as the DSP and PLN risk calculators.
Arrhythmogenic cardiomyopathy (ACM) is a heart muscle disease characterized by replacement of ventricular myocardial by fibrofatty scar tissue which underlies morpho-functional ventricular abnormalities and life-threatening ventricular arrhythmias potentially responsible of suddencardiacdeath (SCD), mostly in young people and athletes 1,2.
There has been a recent surge of interest in efforts to refine the risk stratification for suddencardiacdeath (SCD) in children and adolescents with hypertrophic cardiomyopathy (HCM).
Hypertrophic cardiomyopathy (HCM) is associated with risk of suddencardiacdeath (SCD). Since approval in late 2012, the subcutaneous implantable cardioverter-defibrillator (SICD) has been used as an alternative to the traditional transvenous ICD (TV-ICD) for SCD prevention in HCM.
Predictive factors of benefit from implantation of an implantable cardioverter-defibrillator (ICD) for primary prophylaxis of suddencardiacdeath in patients with nonischemic cardiomyopathy (NICM) have not been clarified.
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a genetic cardiomyopathy resulting from disordered function of the intercalated discs with mainly arrhythmic manifestation, which can be difficult to diagnose because of its variable presentation. The incidence of ARVC is reported to be 1:2000 to 1:5000.
Abstract Cardiomyopathies represent significant contributors to cardiovascular morbidity and mortality. Furthermore, medications targeting core disease processes and/or their downstream adverse effects have been introduced for several cardiomyopathies.
BACKGROUND:Over the past decades, hypertrophic cardiomyopathy has become a contemporary treatable disease. Secondary outcomes included trends of suddencardiacdeath, appropriate/inappropriate shocks, and ICD-related complications.RESULTS:In total, 234 studies (N=92 500, 514 748 patient-years) met inclusion criteria.
Frequent premature ventricular contractions (PVCs) are linked to development of cardiomyopathy (CM) characterized by reduced LV ejection fraction and increased risk of suddencardiacdeath. PVC-CM is thought to be triggered by elevated mechanical myocardial strain due to dyssynchronous contraction during PVCs.
Hypertrophic cardiomyopathy (HCM) is associated with risk of suddencardiacdeath (SCD). Since approval in late 2012, the subcutaneous implantable cardioverter-defibrillator (SICD) has been used as an alternative to the traditional transvenous ICD (TV-ICD) for SCD prevention in HCM.
Hypertrophic cardiomyopathy (HCM) has a prevalence of 1 in 500 people and highly increases the risk of suddencardiacdeath (SCD). Diagnosis is typically based on the Echocardiogram, while cardiac MRI is used for detection of myocardial scarring through Late Gadolinium Enhancement (LGE), an important risk marker for SCD.
BACKGROUND:No disease-specific therapy currently exists for arrhythmogenic right ventricular cardiomyopathy (ARVC), a progressive cardiogenetic condition conferring elevated risk for ventricular arrhythmias, heart failure, and suddencardiacdeath. Emerging gene therapies have the potential to fill this gap.
Researchers Elizabeth Jordan and Ray Hershberger, MD discuss the results of genetic tests that indicate a risk for dilated cardiomyopathy. Dilated cardiomyopathy can be a silent killer DCM is a condition in which the heart muscle weakens and the left ventricle enlarges. The Dilated Cardiomyopathy Consortium was funded by a $12.4
Hypertrophic cardiomyopathy (HCM)-related suddencardiacdeath (SCD) rates with contemporary management are low; however, high-intensity exercise can induce fatal arrhythmias in HCM patients. Thus, current guidelines recommend avoiding high-intensity exercise in HCM patients at high risk for SCD1,2.
BackgroundArrhythmogenic cardiomyopathy (ACM) is a genetic heart muscle disease, which presents with arrhythmias and suddencardiacdeath, along with progressive cardiac remodeling and myocardial inflammation. Journal of the American Heart Association, Ahead of Print.
Dilated cardiomyopathy with arrhythmic phenotype. Abstract Aims Dilated cardiomyopathy (DCM) with arrhythmic phenotype combines phenotypical aspects of DCM and predisposition to ventricular arrhythmias, typical of arrhythmogenic cardiomyopathy.
Hypertrophic cardiomyopathy (HCM) is the leading cause of suddencardiacdeath (SCD) in youth. Cardiac; magnetic resonance imaging (CMR) has been invaluable in improving the risk stratification of these patients for SCD1.
A team at the Centro Nacional de Investigaciones Cardiovasculares (CNIC) has developed an innovative gene-therapy strategy that could transform the treatment of arrhythmogenic right ventricular cardiomyopathy type 5 (ARVC5), a rare and highly penetrant inherited cardiac disorder with an elevated risk of suddencardiacdeath.
BACKGROUND:Hypertrophic cardiomyopathy (HCM) is an important cause of suddencardiacdeath associated with heterogeneous phenotypes, but there is no systematic framework for classifying morphology or assessing associated risks. Circulation: Genomic and Precision Medicine, Ahead of Print.
We organize all of the trending information in your field so you don't have to. Join thousands of users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.



































Let's personalize your content